
Projects
Multiphysical Single-Protein Identification
Deep Learning for Multiphysical Single-Protein Identification
PI: Michael Roukes (Division of Physics, Mathematics and Astronomy)
SASE: Alfredo Gomez, Scholar
The proteomic complexity in biological samples is immense. Within an individual mammalian cell there are roughly 3B proteins, comprising about 10,000 unique types, with concentrations spanning 8 orders of magnitude. These analytes range from cellular proteins represented only by a few copies to those with cellular expression levels of order 100M fold. In human blood serum this range is a thousand-fold greater, spanning 10 orders. It is increasingly clear that resolving the full spectrum of protein constituents present within biological samples is essential for elucidating important, fundamental questions in both fundamental biology and medicine. Carrying out detailed surveys – at the population level, yet with single-molecule resolution – will make it possible to stratify fine details lost in existing consensus-type approaches to proteomics that “average over” the entire population and provide information only about the most prevalent species. Accordingly, single-molecule resolution of proteins and protein complexes in the presence of an immense overabundance of the most prevalent species is required to permit deep proteomic analyses. Surprisingly, no technology exists to achieve this – but we have identified a path that can potentially address both the diversity and the immense numbers of this challenge. We must measure them all!
High-resolution mass spectrometry (MS) is presently the mainstay of proteomics, but it has critical limitations precluding such high-throughput proteomic analysis. These limitations stem from the fact that MS is, at present, a single-channel methodology (providing just one analysis stream) for which Coulomb repulsion strongly limits the number of molecules that can be processed simultaneously. It is projected that these will prevent existing approaches to be sufficiently upscaled to enable deep proteomic profiling of individual cells with extremely high throughput.
The Schmidt Academy worked with the Roukes Group to develop various “data-driven” techniques and algorithms that will enable high-throughput high-resolution mass spectrometry. Year One saw the creation of NEMS-JD (NanoElectroMechanical Systems Jump Detection) and NEMS-FP (NEMS FingerPrint), methods and corresponding python tools designed for measuring the mass of individual molecules using uncharacterized advanced NEMS devices of arbitrary specification, a task that would have otherwise been infeasible.
The Roukes Lab is collaborating with the Schmidt Academy to explore the critical question as to whether a concatenation of “orthogonal” lower-resolution multiphysical analyses, aided by finite element analysis and deep learning, can be melded to achieve a new high-resolution and massively parallel approach to proteomic analysis.
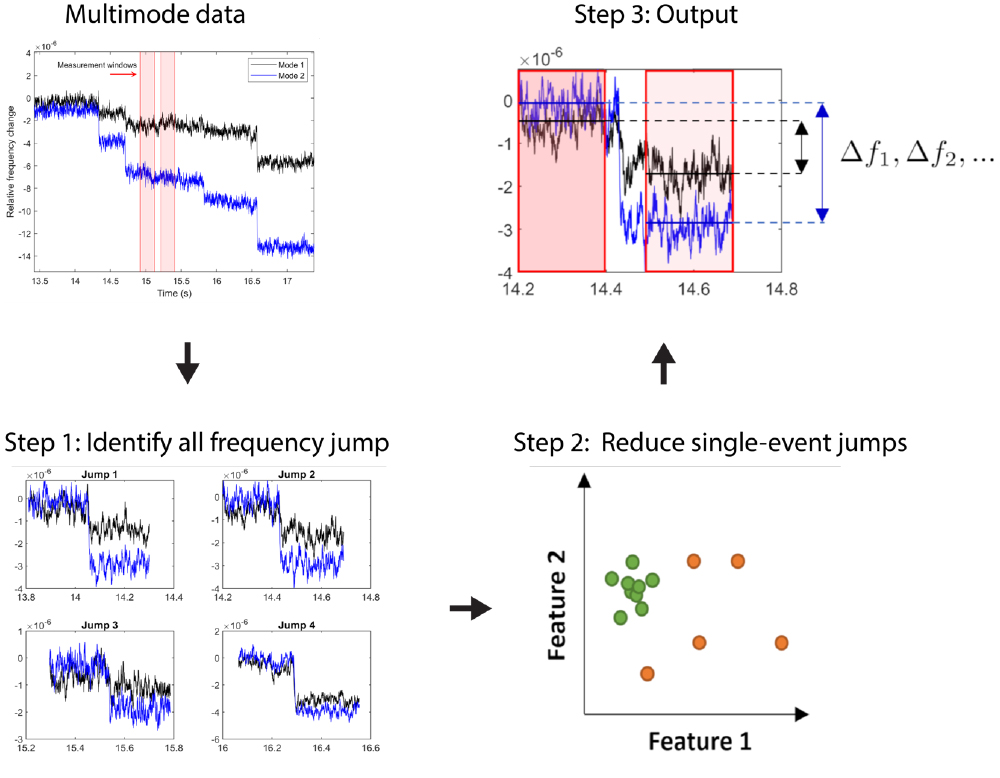
A diagram outlining the algorithm developed for the detection and measurement of NEMS physisorption is shown. After multimodal data has been collected from NEMS instruments, these time series segments are processed and filtered prior to reporting final high-precision measurements.
In year two, the work extended to not only further develop the aforementioned tools, but to also consider different approaches to mass spectrometry such as alternative NEMS designs and ion traps. To that end, deliverables have included drafting new manuscripts1,2 provisional patent disclosures, and various internal software package for simulation and data analysis for these technologies.
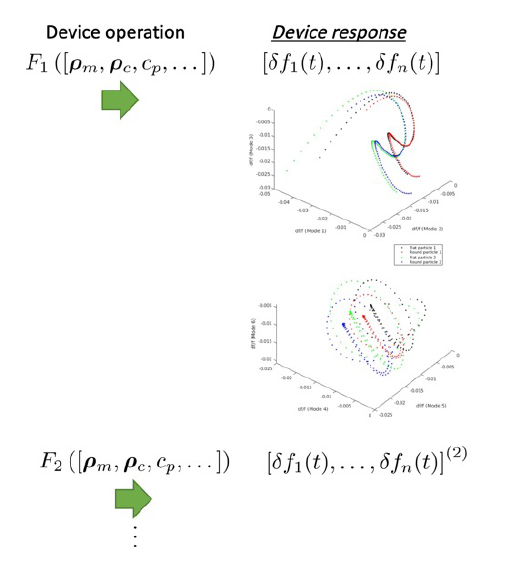
The steady-state change in resonant frequency is shown for the first six vibrational modes for two pairs of particles. Data-driven models can then be employed to differentiate between small changes in these particles by considering slight changes in mass or three-dimensional shape.
1. Neumann, A. P., Gomez, A., Nunn, A. R., Sader, J. E. & Roukes, M. L.Nanomechanical mass measurements through feature-based time series
clustering.
https://pubs.aip.org/aip/rsi/article/95/2/025001/3261868
2. Sader, J. E., Gomez, A., Neumann, A. P., Nunn, A. R., Sader, J. E. &
Roukes, M. L. Data-driven fingerprint nanomechanical mass spectrometry.
https://www.nature.com/articles/s41467-024-51733-8?utm_source=rct_congratemailt&utm_medium=email&utm_campaign=oa_20241022&utm_content=10.1038/s41467-024-51733-8
For a write-up on 2, see:
https://www.caltech.edu/about/news/new-fingerprint-mass-spectrometry-method-paves-the-way-to-solving-the-proteome