
Projects
Deep Learning for Single-cell Proteomics
PI: Michael Roukes (PMA, EAS, and BBE Divisions)
SASE: Andrew Chiang, Scholar
Proteins are the ultimate executors of cellular function. Acting as tiny cellular-level machines, proteins carry out a myriad of biological processes making proteomics, the study of proteins within cells, crucial to understanding the initiation and development of cancer and other disease processes. If realized, single-cell proteomics promises insight into the complex relationships between different proteins and their distribution throughout cells — a breakthrough development for both biology and medicine. Given the fundamental importance of proteomics in biomedicine, it is perhaps surprising that its development has lagged behind the related fields of genomics and transcriptomics. For example, while gene-sequencing for individual cells is now routine, a deep profile of all proteins within a single-cell is yet to be achieved. This disparity can be understood by the immense complexity of proteins in biological samples with a typical mammalian cell containing 3 billion proteins, comprised of over 10,000 distinct types, and concentrations spanning 8 orders of magnitude. This means that while some proteins occur only a handful of times other proteins can have hundreds of millions of copies. Therefore, a deep profile of a single cell necessitates the detection of single proteins in the presence of a staggering overabundance of the most prevalent species.
Caption: Single Cell Proteomics
High resolution mass spectrometers are currently the mainstay of proteomics, however, these instruments lack the sensitivity and throughput required for deep profiling of the single-cell proteome. Our present large-scale effort is overcoming this limitation by developing micro-scale electrostatic traps (μEST) capable of capturing and analyzing individual ions with single-molecule resolution. Through the large-scale integration of thousands of μESTs on a single chip, we will perform massively multiplexed measurements, achieving the sensitivity and high-throughput required to resolve the full spectrum of the single-cell proteome.
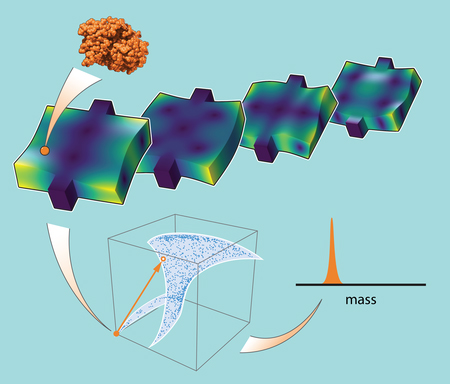
Constructing electrostatic ion traps at the micro-scale is challenging due to several factors. Electrostatic trapping fields deviate from the optimal designs, causing ions to follow complex and chaotic trajectories. To obtain accurate predictions of ion properties, it is necessary to analyze complex dynamics hidden in noisy signals. The Roukes group is developing in-house simulation software and machine learning algorithms to process complex ion signals. The Schmidt Scholar is collaborating closely with our research team to further develop our in-house ion simulation software, including improving and publishing documentation, enhancing meshing routines to handle complex geometries, and assisting with performance upgrades as we integrate fast multipole methods into the software.